| 📰 Title: | Avogadro | 🕹️ / 🛠️ Type: | Game |
|---|---|---|---|
| 🗃️ Genre: | Puzzle | 🚦 Status: | 05. Tested & Working (status) |
| 🏷️ Category: | Puzzle ➤ E-learning ➤ Physics & Chemistry | 🌍️ Browser version: | |
| 🔖 Tags: | Puzzle; E-learning; Chemistry; Plugins system; Flagship | 📦️ Package Name: | avogadro, avogadro-data |
| 🐣️ Approx. start: | 📦️ Arch package: | ||
| 🐓️ Latest: | 2018-09-14 | 📦️ RPM package: | |
| 📍️ Version: | Latest: Avogadro 2 v.1.91.0 / Dev: b89b96c | 📦️ Deb package: | |
| 🏛️ License type: | 🕊️ Libre | 📦️ Flatpak package: | |
| 🏛️ License: | 3-clause BSD | 📦️ AppImage package: | |
| 🏝️ Perspective: | Third person | 📦️ Snap package: | |
| 👁️ Visual: | 3D | ⚙️ Generic binary: | |
| ⏱️ Pacing: | Real Time | 📄️ Source: | |
| 👫️ Played: | Single | 📱️ PDA support: | |
| 🎖️ This record: | 🕳️ Not used: | ||
| 🎀️ Game design: | 👫️ Contrib.: | goupildb & Louis | |
| 🎰️ ID: | 15562 | 🐛️ Created: | 2019-03-12 |
| 🐜️ Updated: | 2022-08-14 |
| [en]: | A libre and multi-platform molecular builder and visualization tool that is part of a software suite called Open Chemistry project. It can visualize properties such as molecular orbitals or electrostatic potentials and features an intuitive molecule builder. It can be used by end-users of the chemical industry and education. It can be extended with C ++ plugins or Python scripts, and integrates with online databases. An intuitive and ergonomic tool. Excellent. | [fr]: | Un outil libre et multi-plateforme de modélisation et d'imagerie moléculaire faisant partie d'une suite logicielle dénommée Open Chemistry. Il permet de visualiser des propriétés comme les orbites moléculaires ou les potentiels électrostatiques et dispose d'un constructeur de molécules intuitif. Il se destine aux utilisateurs finaux de l'industrie chimique et de l'enseignement. Il peut être étendu avec des plugins C ++ ou des scripts Python, et s'intègre aux bases de données en ligne. Un outil intuitif et ergonomique. Excellent. |
|---|
🦉️ From Devs: (202xxx),
🕯️ How To: (202xxx), (202xxx), (202xxx),
🏡️ Website & videos
[Homepage] [Dev site] [Features/About 1 2] [Screenshots] [Videos t(202xxx) ts(202xxx) gd(202xxx) gu(202xxx) id(202xxx) r(202xxx) lp(202xxx) ht(202xxx) ht(202xxx) ht(202xxx) ht(202xxx) ht(202xxx) ht(202xxx) ht(202xxx) g(202xxx) g[fr](202xxx) g[de](202xxx) g[ru](202xxx) g[pl](202xxx) g[cz](202xxx) g[sp](202xxx) ht[pt] g[it](202xxx) g[tr](202xxx)] [WIKI] [FAQ] [RSS] [Changelog 1 2 3]
💰 Commercial: (empty)
🍩️ Resources
• Avogadro 1: [Avogadro 1] [Dev site (GitHub : Eigen 3 compatible) (SourceForge : not compatible with Eigen 3]
(Latest : 1.2.0, 20160608. This version on GitHub compiles well and quickly under Linux: tested.)
🛠️ Technical informations
[Open Hub] [PCGamingWiki] [MobyGames] [Open Chemistry (Building Avogadro 2 and Open Chemistry)] [KitwareBlog (Avogadro 2 and Open Chemistry)]
🐘 Social
Devs (Open Chemistry project [fr] [en]): [Site 1 2] [Chat] [mastodon] [twitter] [PeerTube] [YouTube] [PressKit] [Interview 1(202xxx) 2(202xxx)]
Devs (Kitware [fr] [en]): [Site 1 2] [Chat] [mastodon] [twitter] [Facebook] [PeerTube] [YouTube] [PressKit] [Linkedin] [Interview 1(202xxx) 2(202xxx)]
The Project: [Blog] [Chat] [Forums] [mastodon] [twitter] [Facebook] [PeerTube] [YouTube] [PressKit] [reddit]
🐝️ Related
[Wikipedia (Avogadro) [fr] [en] [de]]
[Debian/Ubuntu]
📦️ Misc. repositories
[Repology] [pkgs.org] [Arch Linux / AUR] [openSUSE] [Debian/Ubuntu] [Flatpak] [AppImage] [Snap] [PortableLinuxGames]
🕵️ Reviews
[HowLongToBeat] [metacritic] [OpenCritic] [iGDB]
📰 News / Source of this Entry (SotE) / News (SotN)
🕊️ Source of this Entry: [Site on Mastodon (date)]
🐘 Social Networking Update (on Mastodon)
🕹️ Title:
🦊️ What's:
🏡️
🐣️
🔖 #Flagship
📦️
📖 Our entry: http://www.lebottindesjeuxlinux.tuxfamily.org/en/online/lights-on/
🥁️ Update:
⚗️
📌️ Changes:
🐘 From:
🏝️ https://invidious.lunar.icu/
🦉️ https://invidious.lunar.icu/
🦝️ https://invidious.lunar.icu/
🦝️ https://invidious.lunar.icu/
🕵️ https://invidious.lunar.icu/
🕯️ https://invidious.lunar.icu/
🕯️ https://invidious.lunar.icu/
🎲️ https://invidious.lunar.icu/
🎲️ https://invidious.lunar.icu/
🎲️[fr] https://invidious.lunar.icu/
🎮️ https://invidious.lunar.icu/
🎮️ https://invidious.lunar.icu/
🐧 https://invidious.lunar.icu/
🐧 https://invidious.lunar.icu/
The Open Chemistry project offers a suite of permissively licensed multi-platform tools that provide reusable libraries and end-user applications for computational chemistry, materials science, and related areas.
Kitware offers services to help you best leverage the Open Chemistry suite. Discover how we can work together to advance your research.
• Avogadro 2 is a versatile desktop application for editing and visualizing molecular data. It is multi-platform, and can be extended with C++ plugins, or Python scripts. Support for a number of computational chemistry codes is offered, and integration with online databases.
• MoleQueue provides desktop integration of high-performance computing (HPC) resources, along with local execution of standalone code. It is a small, Qt-based, system-tray resident application that makes computing resources easier to access from graphical applications.
• Tomviz offers a desktop application for the processing, visualization, and analysis of 3D tomographic data. The full pipeline of data processing steps from alignment and reconstruction right through to analysis and segmentation of the 3D data can be presented, saved, and restored.
Avogadro 2
Introduction
Avogadro is an advanced molecular editor designed for multi-platform use in computational chemistry, molecular modeling, bioinformatics, materials science, and related areas. It offers flexible rendering and a powerful plugin architecture. The code in this repository is a rewrite of Avogadro with source code split across a libraries repository and an application repository.
Core features and goals of the Avogadro project:
 
• Open source distributed under the liberal 3-clause BSD license
• Cross platform with nightly builds on Linux, Mac OS X and Windows
• Intuitive interface designed to be useful to whole community
• Fast and efficient embracing the latest technologies
• Extensible, making extensive use of a plugin architecture
• Flexible supporting a range of chemical data formats and packages
Avogadro 2 is being developed as part of the [Open Chemistry] project at [Kitware], along with companion tools and libraries to support the work. The Avogadro 1.x series currently has more features, and can be found [here]. We will be porting more features to the Avogadro 2 code base, and making regular releases to get feedback from the community.
Installing
We provide nightly binaries built by our [dashboards] for Mac OS X and Windows. If you would like to build from source we recommend that you follow our [building Open Chemistry guide] that will take care of building most dependencies.
Contributing
Our project uses the standard GitHub pull request process for code review and integration. Please check our [development guide] for more details on developing and contributing to the project. The GitHub issue tracker can be used to report bugs, make feature requests, etc.
Our [wiki] is used to document features, flesh out designs and host other documentation. Our API is [documented using Doxygen] with updated documentation generated nightly. We have several [mailing lists] to coordinate development and to provide support.
Debian:
Molecular Graphics and Modelling System
Avogadro is a molecular graphics and modelling system targeted at molecules and biomolecules. It can visualize properties like molecular orbitals or electrostatic potentials and features an intuitive molecular builder.
Features include:
• Molecular modeller with automatic force-field based geometry optimization
• Molecular Mechanics including constraints and conformer searches
• Visualization of molecular orbitals and general isosurfaces
• Visualization of vibrations and plotting of vibrational spectra
• Support for crystallographic unit cells
• Input generation for the Gaussian, GAMESS and MOLPRO quantum chemistry packages
• Flexible plugin architecture and Python scripting
File formats Avogadro can read include PDB, XYZ, CML, CIF, Molden, as well as Gaussian, GAMESS and MOLPRO output.
Wikipedia:
Avogadro is a molecule editor and visualizer designed for cross-platform use in computational chemistry, molecular modeling, bioinformatics, materials science, and related areas. It is extensible via a plugin architecture.
Features
• Molecule builder-editor for Windows, Linux, Unix, and macOS.
• All source code is licensed under the GNU General Public License (GPL) version 2.
• Supported languages include: Chinese, English, French, German, Italian, Russian, Spanish, and Polish.
• Supports multi-threaded rendering and computation.
• Plugin architecture for developers, including rendering, interactive tools, commands, and Python scripts.
• OpenBabel import of files, input generation for multiple computational chemistry packages, X-ray crystallography, and biomolecules.
Un outil de modélisation et d'imagerie moléculaire, par le studio Kitware, dans un projet plus global dénommé l'Open Chemistry project.
Avogadro est un outil libre et multi-plateforme de modélisation et d'imagerie moléculaire faisant partie d'une suite logicielle dénommée Open Chemistry. Il permet de visualiser des propriétés comme les orbites moléculaires ou les potentiels électrostatiques et dispose d'un constructeur de molécules intuitif. Il se destine aux utilisateurs finaux de l'industrie chimique et de l'enseignement. Il peut être étendu avec des plugins C ++ ou des scripts Python, et s'intègre aux bases de données en ligne. Un outil intuitif et ergonomique. Excellent.
Le projet Open Chemistry offre une suite d’outils multi-plateformes sous licence permissive qui fournissent des bibliothèques réutilisables et des applications pour utilisateurs finaux pour la chimie informatique, la science des matériaux et les domaines connexes.
Kitware propose des services pour vous aider à tirer le meilleur parti de la suite Open Chemistry. Découvrez comment nous pouvons travailler ensemble pour faire avancer vos recherches.
• Avogadro 2 est une application de bureau polyvalente permettant d’éditer et de visualiser des données moléculaires. Il est multi-plateforme et peut être étendu avec des plugins C ++ ou des scripts Python. La prise en charge d'un certain nombre de codes de chimie computationnelle est proposée, ainsi que l'intégration aux bases de données en ligne.
• MoleQueue permet l'intégration de ressources de calcul haute performance (HPC) sur le bureau, ainsi que l'exécution locale de code autonome. Il s’agit d’une petite application résidente basée sur Qt et résidant dans la barre système, qui facilite l’accès aux ressources informatiques à partir d’applications graphiques.
• Tomviz propose une application de bureau pour le traitement, la visualisation et l'analyse de données tomographiques 3D. L'ensemble des étapes de traitement des données, depuis l'alignement et la reconstruction jusqu'à l'analyse et la segmentation des données 3D, peut être présenté, enregistré et restauré.
Avogadro 2
Introduction
Avogadro est un éditeur moléculaire avancé conçu pour une utilisation multi-plateforme dans les domaines de la chimie informatique, de la modélisation moléculaire, de la bio-informatique, de la science des matériaux et de domaines connexes. Il offre un rendu flexible et une architecture de plugin puissante. Le code de ce référentiel est une réécriture d’Avogadro avec un code source divisé en un référentiel de bibliothèques et un référentiel d’applications.
Caractéristiques principales et objectifs du projet Avogadro:

• Open source distribué sous la licence libre 3-clause BSD
• multi-plateforme avec des compilations nocturnes sur Linux, Mac OS X et Windows
• Interface intuitive conçue pour être utile à toute la communauté
• Rapide et efficace, intégrant les dernières technologies
• Extensible, faisant largement appel à une architecture de plugin
• Flexible prenant en charge une gamme de formats et de packages de données chimiques
Avogadro 2 est en cours de développement dans le cadre du projet [Open Chemistry] du studio [Kitware], accompagné d'outils et de bibliothèques associés au projet. La série Avogadro 1.x comporte actuellement plus de fonctionnalités et se trouve [ici]. Nous allons porter plus de fonctionnalités dans la base de code Avogadro 2 et publier régulièrement des versions pour obtenir les commentaires de la communauté.
L'installation
Nous fournissons des fichiers binaires nocturnes créés par nos [dashboards] pour Mac OS X et Windows. Si vous souhaitez le construire à partir du source, nous vous recommandons de suivre notre guide [Building Open Chemistry] qui prendra soin de la construction de la plupart des dépendances.
Contribuer
Notre projet utilise le processus standard de pull request GitHub pour la révision et l'intégration de code. Veuillez consulter notre [development guide] pour plus de détails sur le développement et la contribution au projet. L'issue tracker sur GitHub peut être utilisé pour signaler des bugs, faire des demandes de fonctionnalités, etc.
Notre a href=https://wiki.openchemistry.org/Main_Page target=_blank>[wiki] est utilisé pour documenter des fonctionnalités, étoffer des conceptions et héberger d'autres documents. Notre API est [documentée avec Doxygen] avec une documentation mise à jour générée tous les soirs. Nous disposons de plusieurs [mailing lists] pour coordonner le développement et fournir un support.
Debian:
Système de modélisation et d'imagerie moléculaire
Avogadro est un système de modélisation et d'imagerie moléculaire visant les molécules et les biomolécules. Il permet de visualiser des propriétés comme les orbites moléculaires ou les potentiels électrostatiques et dispose d'un constructeur de molécules intuitif.
Fonctions disponibles :
• modeleur moléculaire avec optimisation géométrique basée sur les champs de force automatique ;
• mécanique moléculaire incluant des recherches de contraintes et de conformation ;
• visualisation des orbites moléculaires et d'isosurfaces générales ;
• visualisation de vibrations et graphique du spectre de vibration ;
• gestion des cellules cristallographiques ;
• création d'entrée pour des paquets chimiques Gaussian, GAMESS et MOLPRO ;
• architecture de greffon flexible et script Python.
Les formats de fichiers qu'Avogadro peut lire incluent PDB, XYZ, CML, CIF, Molden ainsi que les sorties Gaussian, GAMESS et MOLPRO.
Wikipedia:
Avogadro est un éditeur de molécule multi-plateforme extensible (grâce à des plugins) et libre, utilisé pour la chimie numérique, la modélisation moléculaire, la bio-informatique, la science des matériaux et les domaines proches.
Caractéristiques
• Éditeur de molécules pour Windows, Linux et Mac OS X.
• Code source disponible sous Licence publique générale GNU.
• Existe en anglais, chinois, allemand, italien, russe et espagnol.
• De multiples plugins existent notamment pour le rendu, les outils interactifs et des scripts Python.
• Peut importer des fichiers OpenBabel.
🔧️ INSTALLATION:
⚙️ Installation à partir du binaire du jeu :
(✔ v. 1.2.0) • L'interface est en dépôt, il suffit d'installer le paquet.
📄️ Installation à partir du source du jeu :
(✔ v. 1.2.0) Avogadro 1 (ne sera plus nécessaire une fois Avogadro 2 terminé) :
Installation :
• Installez au préalable les paquets suivants : # apt install libeigen3-dev libopenbabel-dev sip-dev doxygen libdotconf-dev
• Optionnel : # apt install openbabel openbabel-gui python-openbabel viewmol v-sim-plugins docbook-utils
• Téléchargez son source sur SourceForge GitHub (car il est compatible avec Eigen 3 des dépôts Debian), décompressez-le et placez-vous dans son répertoire racine
• Puis lancez successivement :
$ cmake .
$ make
(ou, pour accélérer la compilation, "$ make -j8" si vous disposez d'un processeur 8 threads, à adapter pour vous)
(✔ v. 1.91.0) Avogadro 2 :
Le source nécessite un certain nombre de dépendances internes au projet et non délivrées par défaut.
Le téléchargement de la version sous forme de container zip ou tar.gz ne suffit pas.
Il est donc nécessaire de télécharger le source via git (procédure décrite ci-après).
Installation :
• Installez au préalable les paquets suivants : # apt install libeigen3-dev libopenbabel-dev sip-dev doxygen
• Optionnel : # apt install openbabel openbabel-gui python-openbabel viewmol v-sim-plugins docbook-utils
• Téléchargez son source via la commande suivante : $ git clone --recursive git://github.com/OpenChemistry/openchemistry.git
• Puis lancez successivement :
$ cd openchemistry
$ mkdir openchemistry-build
$ cd openchemistry-build
$ cmake ..
$ cmake --build . --config Release
(n'oubliez pas le ".")
🚀️ LANCEMENT DE L'INTERFACE:
Avogadro 1
• Si vous avez installé le jeu à partir d'un paquet : Alt F2 puis saisissez : avogadro
• Si vous avez compilé l'interface à partir de son source, en console dans son répertoire racine lancez : $ ./bin/avogadro
Avogadro 2 :
• Si vous avez compilé l'interface à partir de son source, en console dans son répertoire racine (openchemistry) lancez : $ ./openchemistry-build/prefix/bin/avogadro2
🕵️ Test (Avogadro 1 v.1.2.0) par goupildb (config. : Debian Sid 64-bit):
🎯️ Objectif de ce test: tester son fonctionnement.
J'ai d'abord testé Avogadro 1, qui se compile rapidement et facilement, et qui semble (au 1er abord) plus complet (mais Avogadro 2 est bien plus évolué qu'il n'y paraît, du fait de son excellente ergonomie). Le tout pour m'apercevoir qu'il était dans les dépôts Debian et que la version des dépôts fonctionne bien :))
Avogadro 2 est en cours de développement (ré-écriture plus propre) et incorporera à terme les fonctionnalités d'Avogadro 1 (qui est pour l'instant plus complet).
🕵️ Test (Avogadro 2 v.1.91) par goupildb (config. : Debian Sid 64-bit):
🎯️ Objectif de ce test: rédiger sa notice d'installation, tester son fonctionnement et partager mes premières impressions.
• 🫕️ Installation :
Pas de binaire disponible.
La compilation est un peu plus compliquée qu'Avogadro 1, mais elle reste assez simple à lancer (il suffit de suivre la notice).
Néanmoins c'est la plus élaborée qu'il m'ait été de voir jusqu'ici : elle lance une succession de téléchargements (toutes les dépendances nécessaires), de vérifications et de compilations. Un petit bijou d'horlogerie suisse.
Au total la compilation (non parallélisée) aura duré (téléchargements intermédiaires compris) environ 40 minutes.
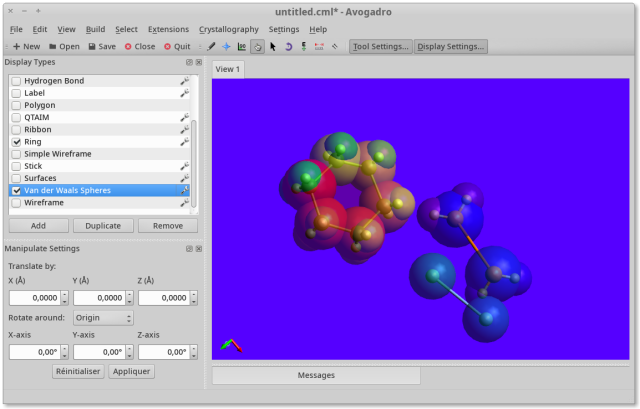
L'interface fonctionne bien (voir la copie d'écran ci-dessus).
Pour la copie d'écran j'ai fait un peu n'importe quoi pour avoir quelque-chose de visuel.
L'équivalent du menu "Settings" (permettant d'ajouter d'autres barres d'outils) d'Avogadro 1 s'obtient par un clic droit sur le haut de l'interface, là où se trouvent les boutons.
À noter que lorsque l'on laisse la souris au dessus d'un bouton on obtient le libellé / l'aide contextuelle du bouton (très utile lorsque l'on ne sait pas à quoi ils servent).
Pour le reste c'est de l'excellent boulot, l'interface est intuitive et réussie visuellement.
On sélectionne une molécule dans un menu déroulant, et elle s'affiche avec les liaisons simple ou double de manière automatique. Si celles du menu déroulant ne suffisent pas, en bas de ce même déroulant une option "Other..." affiche un tableau périodique des éléments permettant d'obtenir un choix plus conséquent.
On peut disposer différentes molécules puis naviguer (zoom, rotations dans l'espace, étirer les liaisons, ...).
Les possibilités sont énormes (chargement et sauvegarde de molécules dans toutes sortes de formats, import, ...) mais je n'ai clairement pas le niveau pour faire autre chose que de m'amuser à dessiner des molécules à partir d'atomes et les observer dans l'espace.
Pour la copie d'écran :
• J'ai changé le fond noir par défaut (menu "View" puis "Set Background Color") par du violet.
• j'ai dessiné un noyau benzénique (C6H6, cyclohexane) à partir de 6 atomes de carbone, et 2 autres molécules au hasard.
• Puis je clique sur le bouton "Display Settings" afin d'accéder au paramétrage de l'affichage de la molécule.
J'ai sélectionné :
"Ball and Stick" (par défaut, pour afficher les atomes)
"Ring" (colorie le centre du noyau benzénique)
"Van der Waals Spheres" (pour afficher les forces éponymes autour des atomes), puis la petite clé à côté du libellé "Van der Waals Spheres" pour mettre le niveau de transparence à environ 60% et sélectionne "Color by Index".
• Il faut ensuite cliquer sur le bouton en forme de flèche 3D violet ("Autorotation tool") sous le menu "Settings" en haut de l'interface, ce qui permet ensuite d'obtenir le menu "Autorotate Settings" lorsque l'on effectue un clic droit sur le haut de l'interface, et je sélectionne "Autorotate Settings" ce qui fait apparaître le menu correspondant. Je n'ai plus qu'à déplacer légèrement les curseurs des x, y et z pour voir ma molécule effectuer une rotation sur elle-même. On peut bien sûr zoomer à souhait par la molette de la souris : superbe !
• N'oubliez pas d'immortaliser la scène par sa sauvegarde (j'ai testé ensuite son chargement : nickel).
Cette scène animée ne consomme que 10% des ressources de mon processeur (8 thread cumulés), donc il ne me semble pas très gourmand.
• 🕹️ Conclusion :
Impression globale : 👍️
L'outil parfait pour se représenter / visualiser des molécules (au lieu des traditionnels boules en plastique qui s'emboîtent). Mais çà, ça n'est qu'un volet des possibilités de l'outil (je n'ai pas les compétences pour tester le reste).
👏️ ❤️ Un grand bravo et merci à ses auteurs !